> Retour liste des Items 6ème édition - Ancien programme
Artérite à cellules géantes
Introduction :
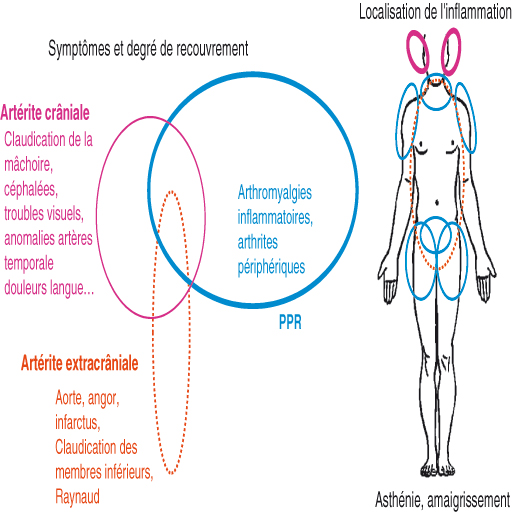
La pseudo-polyarthrite rhizomélique (PPR) et l'artérite à cellules géantes (ACG) ou maladie de Horton sont des affections fréquentes du sujet âgé ayant des liens étroits et seraient des expressions cliniques différentes d'une seule entité physiopathologique.
La maladie de Takayasu est une vascularite rare dont l'expression clinique est distincte.
Pseudo-polyarthrite rhizomélique et artérite à cellules géantes
Ainsi, pour la majorité des auteurs, la PPR et l'ACG sont des expressions cliniques distinctes d'une même maladie, pouvant se succéder l'une à l'autre ou être intriquées et ayant des pronostics évolutifs différents (fig. 17.1).
Fig. 17-1 : Pseudo-polyarthrite rhizomélique (PPR) et artérite à cellules géantes (ACG) ou maladie de Horton.
Les facteurs déclenchants de la PPR et l'ACG ne sont pas connus. Des hypothèses environnementales ont été évoquées du fait du caractère saisonnier des pics d'incidence. Certaines infections virales pourraient en être le déclencheur (parvovirus B19, Mycoplasme pneumoniae), mais il n'y a aucune preuve formelle pour étayer ces hypothèses.
Des facteurs immunogénétiques tels que certains allèles HLA-DR4 seraient associés à la PPR et à l'ACG, mais il existe probablement d'autres facteurs génétiques impliqués, comme par exemple ceux contrôlant la production de protéines de l'inflammation (dont l'interleukine 6 [IL-6]).
Lorsque le mécanisme physiopathologique est déclenché, il entraîne une activation des cellules dendritiques (notamment dans l'ACG) et l'activation préférentielle de plusieurs voies des lymphocytes T (Th-1 et Th-17) avec la production de cytokines inflammatoires (IL-6, IL-12).
L'IL-6 - est un marqueur d'activité clinique de la PPR, c'est également une cytokine exprimée en excès dans l'intima des vaisseaux au cours de l'AGC et dans le sang. Sa production excessive dans ces deux pathologies explique en particulier l'importance du syndrome inflammatoire.
Dans ces deux pathologies, le diagnostic est porté grâce aux symptômes cliniques et à la présence d'un syndrome inflammatoire biologique majeur.
La PPR est un rhumatisme inflammatoire touchant le sujet âgé de plus de 50 ans, se caractérisant cliniquement par des arthromyalgies inflammatoires (douleurs inflammatoires articulaires et des muscles) des ceintures (rhizome désigne la localisation préférentielle à la racine des membres) scapulaires et pelviennes. Elle se manifeste biologiquement par un syndrome inflammatoire.
Le diagnostic de PPR repose sur des éléments clinicobiologiques évocateurs mais non spécifiques. Il est en conséquence toujours nécessaire d'éliminer ses principaux diagnostics différentiels, en particulier la PR du sujet âgé et les rhumatismes microcristallins, qui peuvent également se présenter sous la forme d'une atteinte inflammatoire des ceintures chez le sujet avançant en âge.
De ce fait la PPR est parfois décrite comme un syndrome clinique ou a contrario comme une PPR maladie, selon que l'on parle de la présentation clinique (non spécifique) ou de l'entité physiopathologique.
La constatation d'un tableau clinique compatible avec une PPR doit systématiquement faire rechercher cliniquement une ACG associée.
La PPR associe chez un patient de plus de 50 ans, après un début parfois insidieux :
Il n'y a pas d'examens d'imagerie révélant des signes pathognomoniques de la PPR.
Il n'y a pas de destruction articulaire dans la PPR.
Elle peut révéler des atteintes de type bursite sub-acromio-deltoïdienne bilatérale ou de bursite trochantérienne, de ténosynovite du long biceps, des épanchements et des synovites glénohumérales ou coxofémorales. C'est surtout le caractère bilatéral de ces anomalies et leur caractère inflammatoire en mode Doppler puissance qui orientent vers une PPR, dans la mesure où ces anomalies morphologiques sont isolément fréquentes chez le sujet avançant en âge.
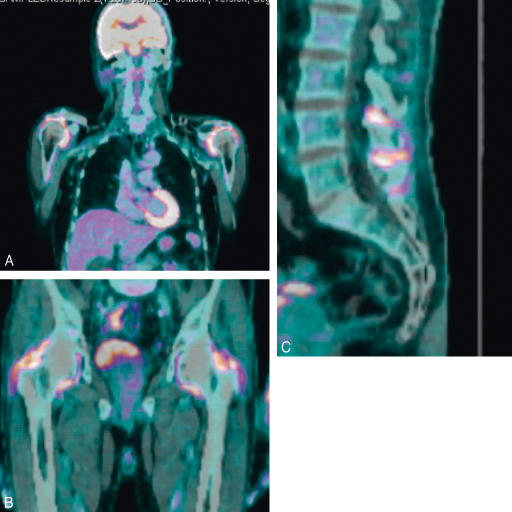
Cet examen avec un marquage au 18-fluoro-désoxy-glucose (18-FDG) permet de visualiser les zones métaboliquement actives consommant du glucose. Il existe dans la PPR des fixations très importantes et bilatérales (même sur les sites asymptomatiques cliniquement) des ceintures scapulaires et pelviennes ainsi que des bursites interépineuses cervicales, dorsales ou lombaires (fig. 17.2). Même si ces fixations sont évocatrices, elles ne sont pas pathognomoniques. Du fait de son coût et de son caractère irradiant, cet examen doit être réservé à une prescription ciblée en cas de doute diagnostique.
Fig. 17-2 : TEP-scan dans la PPR.
A à C. TEP-scan : hyperfixations évocatrices (mais non spécifiques) au niveau des deux épaules, des hanches et bursites interépineuses. Examen à réserver aux diagnostics complexes.
Source : Pr V. Devauchelle-Pensec.
Le diagnostic de PPR repose sur un faisceau d'arguments positifs (arthromyalgies inflammatoires des ceintures associées à un syndrome inflammatoire biologique) et sur des arguments négatifs (absence d'arguments cliniques, biologiques et radiologiques en faveur d'une affection pouvant mimer une PPR).
a. Rhumatismes microcristallins
La chondrocalcinose ou le rhumatisme à hydroxyapatite sont les diagnostics différentiels les plus fréquents de la PPR. Il faut rechercher des liserés calciques articulaires ou abarticulaires sur les radiographies (cf. chapitre 20), des lésions évocatrices en échographie (images en tempête de neige). Le diagnostic est confirmé par la présence de microcristaux de pyrophosphate de calcium ou d'urate de sodium à la ponction articulaire (fréquence des dépôts microcristallins chez le sujet > 50 ans).
b. Polyarthrite rhumatoïde du sujet âgé
Il s'agit d'un diagnostic différentiel difficile car la polyarthrite rhumatoïde du sujet âgé à fréquemment un début rhizomélique (40 %) et la PPR peut comporter des ténosynovites et des arthrites périphériques. Les clichés radiographiques à la recherche d'érosions caractéristiques de la polyarthrite rhumatoïde, ainsi que la recherche d'anticorps anti-peptides citrullinés (ACPA, test anti-CCP) permettront de retenir le diagnostic. Les facteurs rhumatoïdes sont fréquemment élevés sans valeur pathologique dans cette tranche d'âge.
c. RS3PE
La polyarthrite Sdémateuse (ou Remitting Seronegative Symetrical Synovitis with Pitting Edema [RS3PE]) comporte des Sdèmes des extrémités, importants, blancs, prenant le godet et une polysynovite. La polyarthrite Sdémateuse est résolutive généralement en douze à dix-huit mois. Elle est très corticosensible.
d. Spondyloarthrite du sujet âgé
La spondyloarthrite peut se manifester chez le sujet âgé par une atteinte rachidienne et des ceintures faisant évoquer une PPR. Il s'agit néanmoins d'un diagnostic exceptionnel chez le sujet âgé.
a. Affections musculaires inflammatoires
Il s'agit principalement de la polymyosite (recherche de déficit musculaire et d'une élévation des enzymes musculaires et d'autoanticorps) où l'atteinte musculaire est prédominante. Dans la PPR, les enzymes CPK sont normales.
b. Connectivites
Il est possible mais rare qu'un lupus ou un syndrome de Gougerot-Sjögren débute par une atteinte articulaire rhizomélique.
c. Cancers
Un syndrome PPR peut être paranéoplasique. Une forte altération de l'état général ou d'autres signes d'appel généraux doivent faire rechercher des cancers solides ou des lymphomes et hémopathies. Une radiographie pulmonaire est recommandée dans le bilan initial d'une PPR.
d. Infections
Le diagnostic de PPR ne doit être retenu que très exceptionnellement en l'absence de syndrome inflammatoire biologique mais d'autres pathologies peuvent mimer des arthromyalgies et faire errer le diagnostic.
a. Pathologies mécaniques
Les atteintes dégénératives volontiers bilatérales de la coiffe des rotateurs, les lésions arthrosiques coxofémorales ou glénohumérales, les tendinopathies peuvent mimer une PPR.
b. Traitements
Certains médicaments sont susceptibles d'entraîner des myalgies :
c. Autres
Ils sont adaptés en fonction des patients et des signes cliniques.
Tableau 17.1 : Examens complémentaires (liste non exhaustive) à réaliser devant une suspicion de PPR.
| Examens systématiques | En fonction du contexte | Bilan préthérapeutique (hors examens déjà réalisés) | |
|---|---|---|---|
| Biologie | - Hémogramme : - CRP (ou parfois VS) - ionogramme, calcémie - urée, créatininémie et débit de filtration glomérulaire - GGT, ASAT, ALAT - Bandelette urinaire - Anticorps anti-peptides citrullinés (ACPA, anti-CCP) ± facteurs rhumatoïdes | - CPK, kit-myosites - TSH - Électrophorèse des protéines sériques - Anticorps anti-nucléaires, ANCA… | - Glycémie à jeun - Bilan lipidique |
| Imagerie | - Radiographie pulmonaire - Radiographie des épaules (face, rotations interne et externe) - Radiographie du bassin de face | - Échographies des épaules et des hanches - TEP-scan | Ostéodensitométrie |
La maladie de Horton, ou artérite gigantocellulaire (aussi appelée artérite à cellules géantes), est une vascularite systémique primitive correspondant à une panartérite (inflammation atteignant toute la paroi des artères), segmentaire et focale des artères de grand et moyen calibre. Elle peut atteindre tous les vaisseaux de ces calibres, mais préférentiellement et par ordre de fréquence les branches de la carotide externe (artères temporales et occipitales) ainsi que les artères ophtalmiques, vertébrales, sous-claviculaires, axillaires et l'aorte thoracique. On décrit classiquement les formes crâniales qui sont les plus fréquentes et les formes extracrâniales.
C'est la plus fréquente des vascularites après l'âge de cinquante ans.
Elle est fréquemment associée à la PPR.
L'ACG est une urgence diagnostique et thérapeutique du fait des risques d'occlusion des vaisseaux.
Le diagnostic repose sur l'association de symptômes vasculaires, principalement des céphalées et la présence d'un syndrome inflammatoire biologique.
Il est présent chez 15 % des malades et parfois révélateur d'ACG.
L'altération de l'état général et la fièvre sont habituellement plus marquées que dans la pseudo-polyarthrite rhizomélique isolée.
Les symptômes vasculaires oculaires sont à rechercher systématiquement :
Le moindre signe ophtalmologique doit faire redouter une complication ophtalmique grave et définitive et faire discuter une corticothérapie en urgence.
Les complications de l'ACG sont essentiellement vasculaires et ischémiques. Elles sont le plus souvent brutales et irréversibles. Elles font toute la gravité de la maladie et doivent être recherchées systématiquement lors du diagnostic et des consultations de suivi.
a. Complications oculaires
Une complication oculaire survient chez 5 à 20 % des patients et se révèle le plus souvent par une cécité monoculaire brutale pouvant être précédée de prodrome (flou visuel, scotome, diplopie). Un à deux pour cent des patients ont une cécité bilatérale définitive et 2 à 5 % une cécité monoculaire.
L'amaurose est la conséquence :
En cas d'atteinte unilatérale, le risque d'atteinte controlatérale et de cécité totale définitive est important et justifie une prise en charge thérapeutique urgente.
b. Complications neurologiques
Manifestations neuropsychiatriques
Ces manifestations apparaissent dans 3 % des cas avec désorientation temporospatiale et trouble de l'humeur, dont le mécanisme n'est pas totalement éclairci. Ces troubles répondent rapidement à la corticothérapie.
Atteinte neurologique périphérique
Il n'y a pas d'atteinte neurologique périphérique spécifique de l'ACG. Lorsqu'une neuropathie périphérique survient, notamment une mononeuropathie multiple, liée à une atteinte de la vasa nervosum, il faut évoquer une vascularite des vaisseaux de petit calibre qui peut se localiser au niveau de l'artère temporale et se manifester sous la forme de céphalées temporales.
c. Autres complications vasculaires
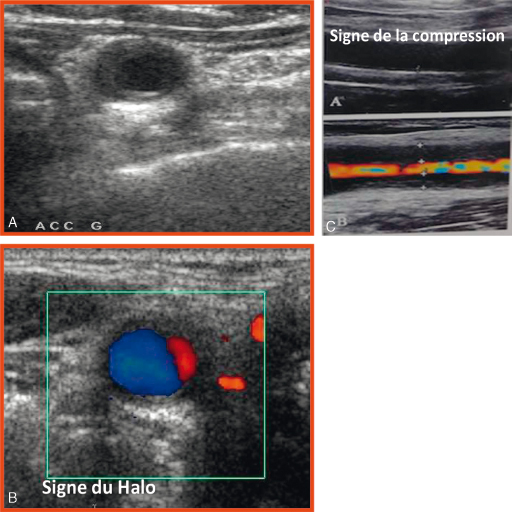
L'échographie des axes vasculaires notamment des artères temporales peut révéler des signes caractéristiques d'artérite avec notamment le signe du halo (halo hypoéchogéne circonférentiel de la paroi du vaisseau), des signes de thrombose ou de sténose artérielle. Elle doit impérativement être réalisée par un opérateur entraîné sachant faire la différence avec des lésions athéromateuses fréquentes à cet âge (fig. 17.3). Cet examen peut donc permettre de confirmer le diagnostic ou de diriger la biopsie de l'artère afin d'améliorer la sensibilité diagnostique de cet examen. Il est recommandé d'étudier les axes vasculaires temporaux mais également les gros vaisseaux du cou et les troncs supra-aortiques.
Fig. 17-3 : Échographie mode B et Doppler des artères temporales, signe du halo.
Échographie mode B et Doppler des artères temporales : artères temporales + vaisseaux du cou et gros troncs thoraciques + artères fémoropoplitées. Lésions : sténose, dissection, occlusion ; signe du halo ++ ; slide sign.
Source : Dr Jousse-Joulin.
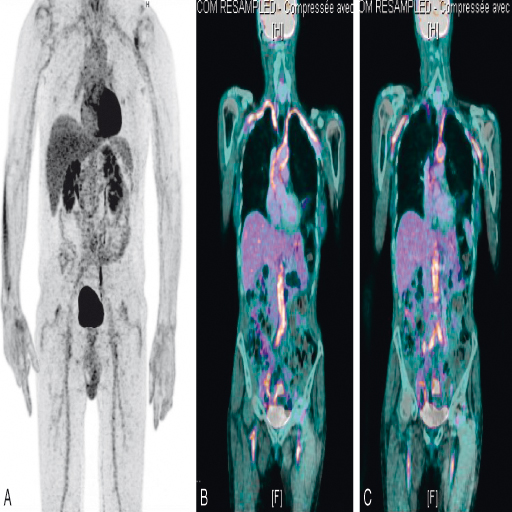
Cet examen marqué au 18-FDG comme dans la PPR, permet de visualiser les atteintes des vaisseaux extracrâniens (fig. 17.4) et d'obtenir une cartographie de l'ensemble des vaisseaux atteints montrant des zones inflammatoires de fixation intense du traceur radioactif utilisé. Les coupes scanographiques associées permettent de visualiser plus précisément l'atteinte de l'aorte. Il est de plus en plus réalisé pour confirmer le diagnostic de la maladie. C'est actuellement un des examens de référence dans l'artérite à cellules géantes.
Fig. 17-4 : TEP-scan dans l'artérite à cellules géantes.
TEP-scan : hyperfixations très évocatrices d'une artérite au niveau des artères axillaires, de l'aorte abdominale et des artères fémorales.
Source : Pr V. Devauchelle-Pensec.
Elle peut permettre de visualiser les atteintes des vaisseaux intracrâniens non accessibles au TEP-scan.
La biopsie de l'artère temporale (BAT) reste l'examen de référence pour confirmer une ACG. L'imagerie de l'artère temporale ou de l'aorte et de ses branches est parfois utilisée en tant que test diagnostique non invasif qui se substitue à la BAT.
La biopsie de l'artère temporale ne doit pas être réalisée devant un tableau de pseudo-polyarthrite rhizomélique isolée typique sans signes cliniques évocateurs d'atteinte vasculaire inflammatoire. La biopsie de l'artère temporale ne doit pas retarder le traitement et elle peut être encore réalisée dans les 14 jours suivant l'instauration de la corticothérapie. Au-delà, les anomalies histologiques se normalisent.
Le caractère focal de l'artérite justifie un prélèvement de 1 cm de long (qui doit être analysé en totalité par le pathologiste). L'examen anatomopathologique met en évidence une panartérite segmentaire et focale. Pour être considérée comme preuve histologique d'une ACG, la biopsie doit nécessairement montrer un infiltrat inflammatoire mononucléé de la média et/ou de l'intima. La présence surajoutée d'une élastophagie de la limitante élastique interne et/ou de cellules géantes est pathognomonique mais inconstante.
La négativité de la biopsie de l'artère temporale n'exclut pas le diagnostic d'ACG. Le caractère focal de la maladie explique la fréquence des faux négatifs (sensibilité de la biopsie de l'artère temporale : entre 50 et 91 %).
Elles sont plus rares à cet âge (maladie de Takayasu, qui atteint le sujet jeune, granulomatose avec polyangéite (ex-maladie de Wegener), périartérite noueuse, maladie de Behcet).
a. Règles hygiénodiététique
b. Autres
La décroissance progressive n'est proposée qu'après la disparition des signes cliniques et la régression du syndrome inflammatoire biologique.
L'absence de corticosensibilité doit remettre en cause le diagnostic.
Le rationnel de l'utilisation du méthotrexate dans cette pathologie est celui d'une épargne cortisonique chez les patients corticodépendants. Il n'y a cependant aucune preuve scientifique définitive de l'efficacité du méthotrexate dans cette indication.
Le tocilizumab (anticorps monoclonal anti-IL-6 - récepteur) est efficace dans le traitement de l'ACG et de la PPR. Il a obtenu une autorisation de mise sur le marché en 2017 dans l'ACG. Sa prescription est réservée actuellement aux spécialistes du domaine, plutôt dans un but d'épargne cortisonique chez des patients ayant des comorbidités (ex. : ostéoporose fracturaire sévère, diabète décompensé, troubles psychiatriques).
Maladie de Takayasu
La maladie de Takayasu est une vascularite des gros vaisseaux, atteignant préférentiellement l'aorte et ses gros troncs, ainsi que l'artère pulmonaire.
Elle peut atteindre tous les gros vaisseaux mais dans 60 à 90 % des cas, il s'agit de l'aorte ascendante ou descendante, des artères sub-claviculaires et des branches de la carotide.
Elle était communément appelée « la maladie des femmes sans pouls » ou le « syndrome de l'arc aortique ».
Celle-ci est inconnue mais deux hypothèses principales sont classiquement avancées :
La maladie de Takayasu a été initialement décrite en Asie où elle est plus fréquente mais elle est universellement répandue.
Elle atteint préférentiellement les femmes (70 %) jeunes (20 à 35 ans) et peut débuter dans l'enfance.
C'est une maladie rare dont l'incidence annuelle est estimée à 2-3/1 000 000 habitants.
Son phénotype semble différent en fonction de l'ethnie et du sexe avec un diagnostic qui serait plus tardif chez les sujets caucasiens (formes moins sévères) et les hommes qui présenteraient plus d'atteinte de l'aorte descendante et abdominale.
La maladie évolue habituellement selon deux phases.
Elle peut associer :
Cette phase dure de quelques semaines à quelques mois. Elle peut récidiver ou disparaître avec un temps de latence de plusieurs années avant la survenue de la phase occlusive.
Les signes cliniques sont liés à la sténose, l'occlusion ou l'anévrisme des artères et varient selon le territoire vasculaire atteint. Des formes longtemps indolentes ou avec des signes peu spécifiques peuvent aboutir à des retards de diagnostic.
Les signes les plus fréquents sont l'apparition d'une HTA, d'une asymétrie tensionnelle, la disparition d'un pouls (notamment huméral), la claudication d'un membre supérieur chez un sujet de moins de 40 ans (syndrome de l'arc aortique) :
Les accidents vasculaires cérébraux et les infarctus du myocarde sont rares.
Les grossesses nécessitent une attention particulière car elles peuvent entraîner plus de complications chez les femmes jeunes en âge de procréer (prématurités, hypertension artérielle maternelle, pré-éclampsie, morts foetales, complications liées aux traitements).
Il n'y a aucun marqueur sérologique spécifique.
Le syndrome inflammatoire (VS ou CRP) est non spécifique mais évocateur d'une artérite. Il peut être absent même en cas de maladie active.
La confirmation diagnostique se fait grâce à l'imagerie. Actuellement ce sont l'échographie-Doppler artérielle (par des opérateurs très entraînés), l'IRM et le TEP-scan qui sont au premier plan pour la confirmation diagnostique. L'angioscanner est moins fréquemment utilisé.
La biopsie artérielle montre des lésions inflammatoires multifocales et segmentaires touchant les trois tuniques. Elle peut être aspécifique ou montrer des lésions cicatricielles.
Du fait des progrès de l'imagerie, le diagnostic histologique est de moins en moins nécessaire au diagnostic.
Maladie rare pouvant bénéficier d'une prise en charge en ALD.
Il repose sur la corticothérapie prescrite initialement à la posologie de 1 mg/kg/j pendant 2 à 4 semaines. Ensuite, en fonction de l'évolution, la posologie est diminuée progressivement pour une durée totale du traitement de 12 à 24 mois. Ce traitement est efficace dans plus de la moitié des cas.
Dans les formes rebelles ou nécessitant de maintenir des doses de cortisone élevées, l'ajout d'un traitement immunosuppresseur est nécessaire : méthotrexate (20-25 mg par semaine), azathioprine, mycophénolate mofétil ou, plus rarement, cyclophosphamide. Enfin, dans des formes graves, des résultats encourageants ont été obtenus avec des biothérapies de type anti-TNF ou par anticorps anti-récepteurs de l'IL-6 - (tocilizumab).
À ces traitements spécifiques, sont associés les traitements médicamenteux classiques et les mesures hygiénodiététiques visant à prévenir les complications de la corticothérapie (ostéoporose cortico-induite…) ou de la maladie (traitements contre l'hypertension artérielle).
L'éducation thérapeutique du patient est indispensable.
Pour limiter les conséquences d'une atteinte artérielle, ils permettent de réaliser des pontages artériels, des désobstructions ou la pose de prothèses vasculaires.
Le risque de resténose à long terme est de 30 %.
Le suivi se fait en milieu spécialisé rhumatologique, de médecine vasculaire ou de médecine interne le plus souvent.
La grossesse et l'accouchement impliquent une surveillance particulière.
L'évolution est le plus souvent lente et indolente et le pronostic est bon en l'absence d'occlusions.
La survie globale est de 90 % à 10 ans et la surmortalité de 3 % environ dans les études les plus récentes.
La réapparition des pouls, la disparition des douleurs, la normalisation de la tension artérielle et l'absence de complication liées au traitement font partie du suivi.
