Myélome multiple des os
![]() La gammapathie monoclonale de signification indéterminée (MGUS, Monoclonal Gammopathy of Undetermined Significance, pour les Anglo-Saxons) est la plus fréquente des gammapathies monoclonales.
La gammapathie monoclonale de signification indéterminée (MGUS, Monoclonal Gammopathy of Undetermined Significance, pour les Anglo-Saxons) est la plus fréquente des gammapathies monoclonales.
![]() Les patients avec MGUS nécessitent donc d'être suivis régulièrement.
Les patients avec MGUS nécessitent donc d'être suivis régulièrement.![]() L'électrophorèse des protéines sériques (EPS) peut être répétée à 6 mois puis de façon annuelle. La surveillance sera ensuite adaptée en fonction de l'existence ou non de facteurs de risque de progression au diagnostic, de l'évolution de la gammapathie (stabilité ou augmentation progressive).
L'électrophorèse des protéines sériques (EPS) peut être répétée à 6 mois puis de façon annuelle. La surveillance sera ensuite adaptée en fonction de l'existence ou non de facteurs de risque de progression au diagnostic, de l'évolution de la gammapathie (stabilité ou augmentation progressive).
![]() Le myélome multiple est une hémopathie maligne liée à une prolifération clonale de plasmocytes tumoraux dans la moelle osseuse.
Le myélome multiple est une hémopathie maligne liée à une prolifération clonale de plasmocytes tumoraux dans la moelle osseuse.
Une immunoglobuline monoclonale, identifiée par sa chaîne lourde et/ou sa chaîne légère, est synthétisée par un clone de lymphocytes B ; elle est le témoin d’une prolifération lymphocytaire B clonale qui peut être bénigne ou maligne.
Le myélome multiple représente 10 % des hémopathies malignes. C’est une affection du sujet de plus de 50 ans. Son incidence augmente avec l’âge ; l’âge moyen au diagnostic est de 65 ans. Il est légèrement plus fréquent chez l’homme que chez la femme.
Le myélome multiple est une maladie très « polymorphe ». Toutes les disciplines médicales peuvent être confrontées aux manifestations d’un myélome non connu :
Le diagnostic repose sur :
Fig. 28.1 :![]() Électrophorèse des protéines sériques.
Électrophorèse des protéines sériques.
A. EPS normale. B. EPS révélant un pic monoclonal dans les gammaglobulines.
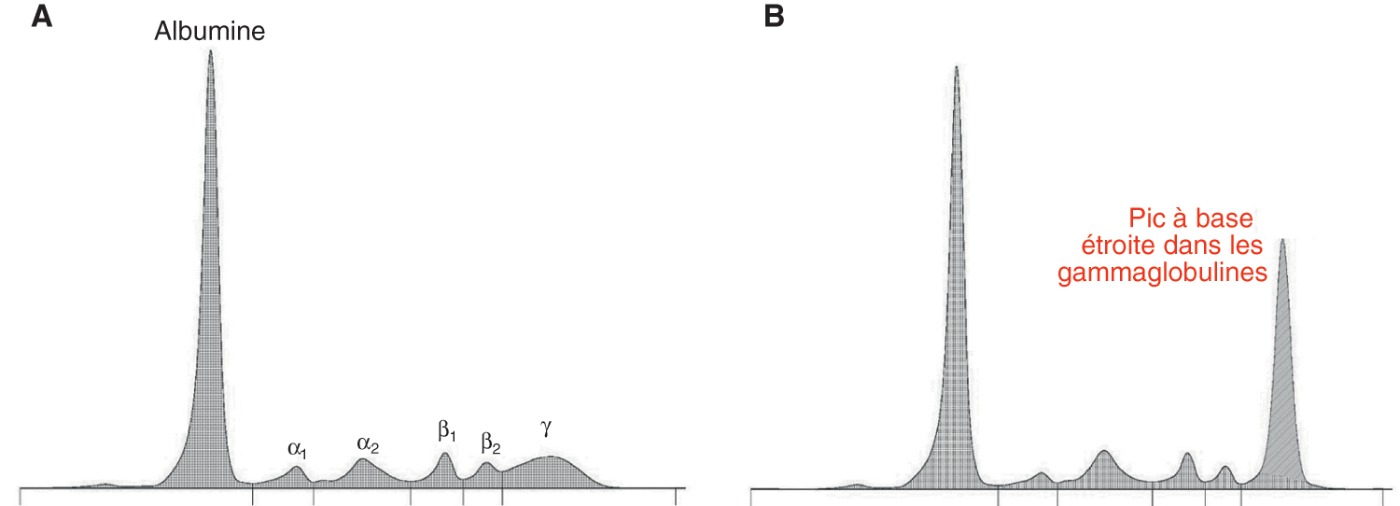
Hypergammaglobulinémie monoclonale (fig. 28.1B) : présence d’un pic à base étroite et symétrique qui migre dans la zone des γ-globulines lorsqu’il s’agit d’une IgG ou, plus rarement, dans la zone des β-globulines lorsqu’il s’agit d’une IgA.
En cas de myélome à chaînes légères, il faut rechercher une hypogammaglobulinémie. La chaîne légère est éliminée le plus souvent dans les urines et donc non visible sur l’EPS (BU normale et protéinurie positive).
La clonalité doit être confirmée par l’immunoélectrophorèse (IgG >> IgA > IgM, ou plus rarement IgD, 1 %, ou IgE, exceptionnelle).
Il a pour but d'apprécier :
![]() Le myélogramme doit être complété par l'analyse caryotypique des plasmocytes médullaires qui a un intérêt pronostique.
Le myélogramme doit être complété par l'analyse caryotypique des plasmocytes médullaires qui a un intérêt pronostique.![]() Si, exceptionnellement, le myélogramme est normal, la biopsie ostéomédullaire au niveau de l'épine iliaque postérosupérieure doit être réalisée afin de confirmer le diagnostic.
Si, exceptionnellement, le myélogramme est normal, la biopsie ostéomédullaire au niveau de l'épine iliaque postérosupérieure doit être réalisée afin de confirmer le diagnostic.
Le caractère symptomatique du myélome dont dépend l’indication du traitement repose sur l’existence de symptômes cliniques ou d’une atteinte d’organe définie par au moins une des anomalies suivantes (critères CRAB) (tableau 28.1) :
Tableau 28.1 : ![]() Définition des formes cliniques (HAS, 2010 ; IMWG, 2014).
Définition des formes cliniques (HAS, 2010 ; IMWG, 2014).
| MGUS | Immunoglobuline monoclonale détectée mais < 30 g/l si IgG et plasmocytose médullaire < 10 % | Pas de symptômes (critères CRAB) |
| Myélome multiple asymptomatique ou indolent | Immunoglobuline monoclonale détectée > 30 g/l et/ou plasmocytose médullaire ≥ 10 % | Pas de symptômes (ni critères CRAB, ni critères de malignité) |
| Myélome multiple symptomatique | Immunoglobuline monoclonale détectée dans le sérum et ou les urines et/ou plasmocytose médullaire ≥ 10 % | Symptômes : – au moins 1 critère CRAB – et/ou 1 critère SLiM CRAB |
Trois critères de malignité SLiM CRAB ont été ajoutés aux critères CRAB et définissent également le caractère symptomatique du myélome. Il s'agit de :
Les examens nécessaires pour mettre en évidence ce composant monoclonal sont :
La découverte d'un pic monoclonal nécessite la réalisation d'un bilan minimal :
![]() Les manifestations osseuses résultent d'une augmentation de l'activité ostéoclastique au contact des plasmocytes : augmentation de la résorption osseuse avec ostéolyse diffuse ou multifocale (fig. 28.2).
Les manifestations osseuses résultent d'une augmentation de l'activité ostéoclastique au contact des plasmocytes : augmentation de la résorption osseuse avec ostéolyse diffuse ou multifocale (fig. 28.2).
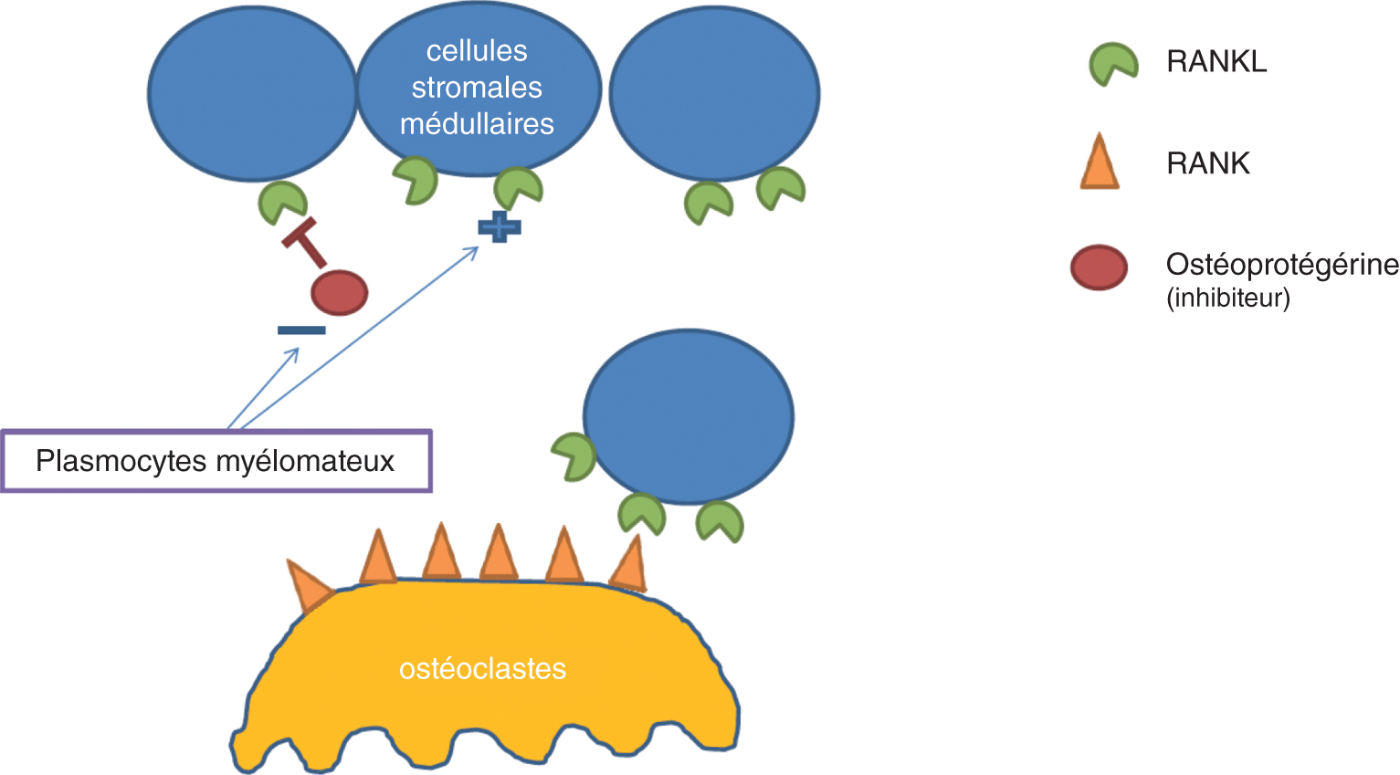
Fig. 28.2 : ![]() Système RANK-RANKL-OPG et myélome.
Système RANK-RANKL-OPG et myélome.
Le plasmocyte myélomateux induit une augmentation de RANKL et une baisse de l’ostéoprotégerine (OPG), régulateur négatif du système. Cela aboutit à la stimulation de l’ostéoclastose.
Source : Inserm 1184.
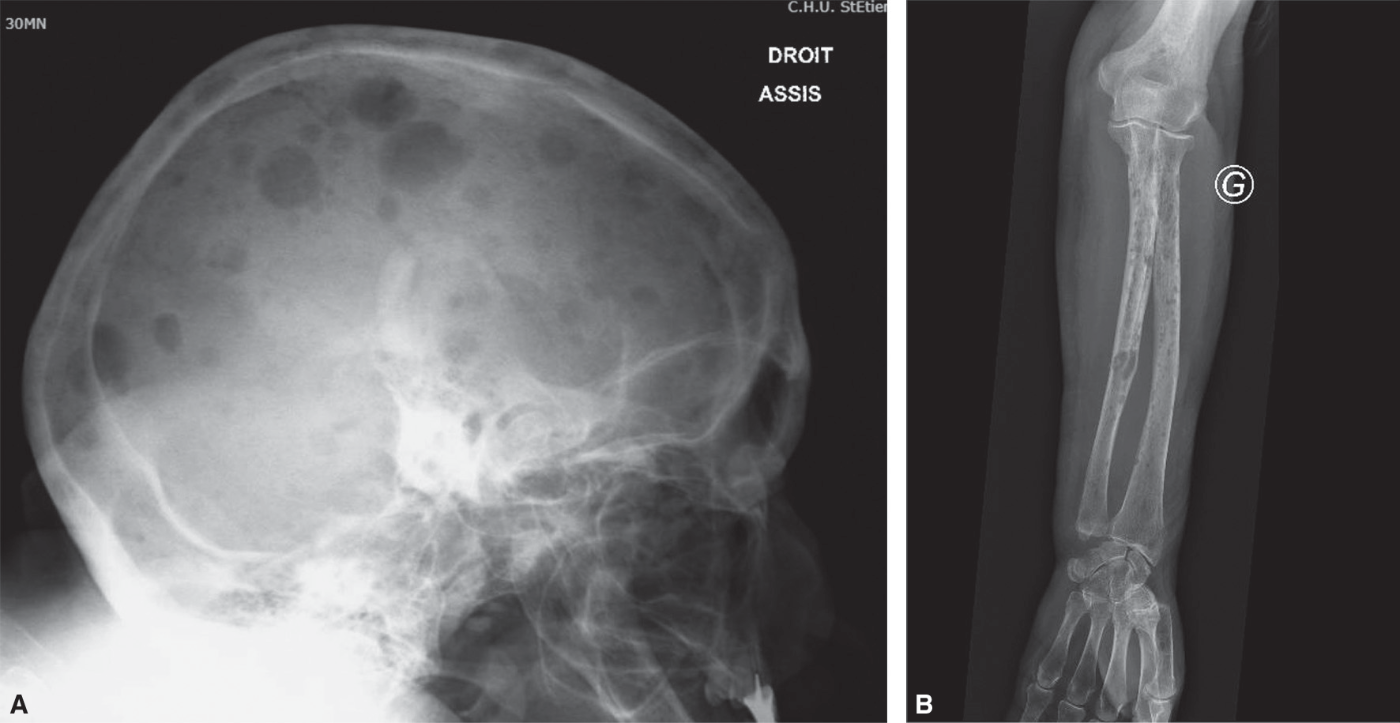
![]() Le myélome est responsable de lésion lytique de type lacune « à l’emporte-pièce », ovalaire ou ronde, sans structure interne visible, bien limitée, sans réaction périphérique ; elle est multiple dans 80 % des cas et tous les os peuvent être atteints (fig. 28.3). Il est parfois difficile de différencier fracture vertébrale d’origine ostéoporotique et fracture liée au myélome. Il est donc nécessaire de toujours réaliser une EPP en cas de fracture ver-tébrale même en cas d’aspect radiologique évocateur de fracture ostéoporotique.
Le myélome est responsable de lésion lytique de type lacune « à l’emporte-pièce », ovalaire ou ronde, sans structure interne visible, bien limitée, sans réaction périphérique ; elle est multiple dans 80 % des cas et tous les os peuvent être atteints (fig. 28.3). Il est parfois difficile de différencier fracture vertébrale d’origine ostéoporotique et fracture liée au myélome. Il est donc nécessaire de toujours réaliser une EPP en cas de fracture ver-tébrale même en cas d’aspect radiologique évocateur de fracture ostéoporotique.
Fig. 28.3 :![]() Aspect radiographique
Aspect radiographique
A. Lacunes multiples « à l’emporte-pièce » du crâne. B. Lésions lytiques « à l’emporte-pièce » des os de l’avant-bras.
L’IRM panrachidienne et du bassin est de plus en plus pratiquée lorsque le diagnostic de myélome est établi. Elle permet d’analyser la moelle osseuse à la recherche d’une infiltration médullaire en l’absence de lésions sur les radiographies, notamment dans le myélome asymptomatique (= indolent). L’aspect typique de cette infiltration médullaire est décrit comme étant « poivre et sel ».
Elle permet en cas de symptomatologie douloureuse rachidienne de rechercher une épidurite, une compression radiculaire ou médullaire.
Le scanner faible dose (low dose) « corps entier » a remplacé les radiographies du squelette dans le bilan initial car plus sensible et spécifique.
L’injection de produit de contraste iodé n’est pas indiquée dans le bilan radiologique. Elle est à éviter du fait du risque d’atteinte rénale.
Fig. 28.4 :![]() Image lytique en TDM avec lésion lytique de L4 touchant l’arc postérieur.
Image lytique en TDM avec lésion lytique de L4 touchant l’arc postérieur.
A. Coupe coronale. B. Coupe horizontale. C. Lésion costale.

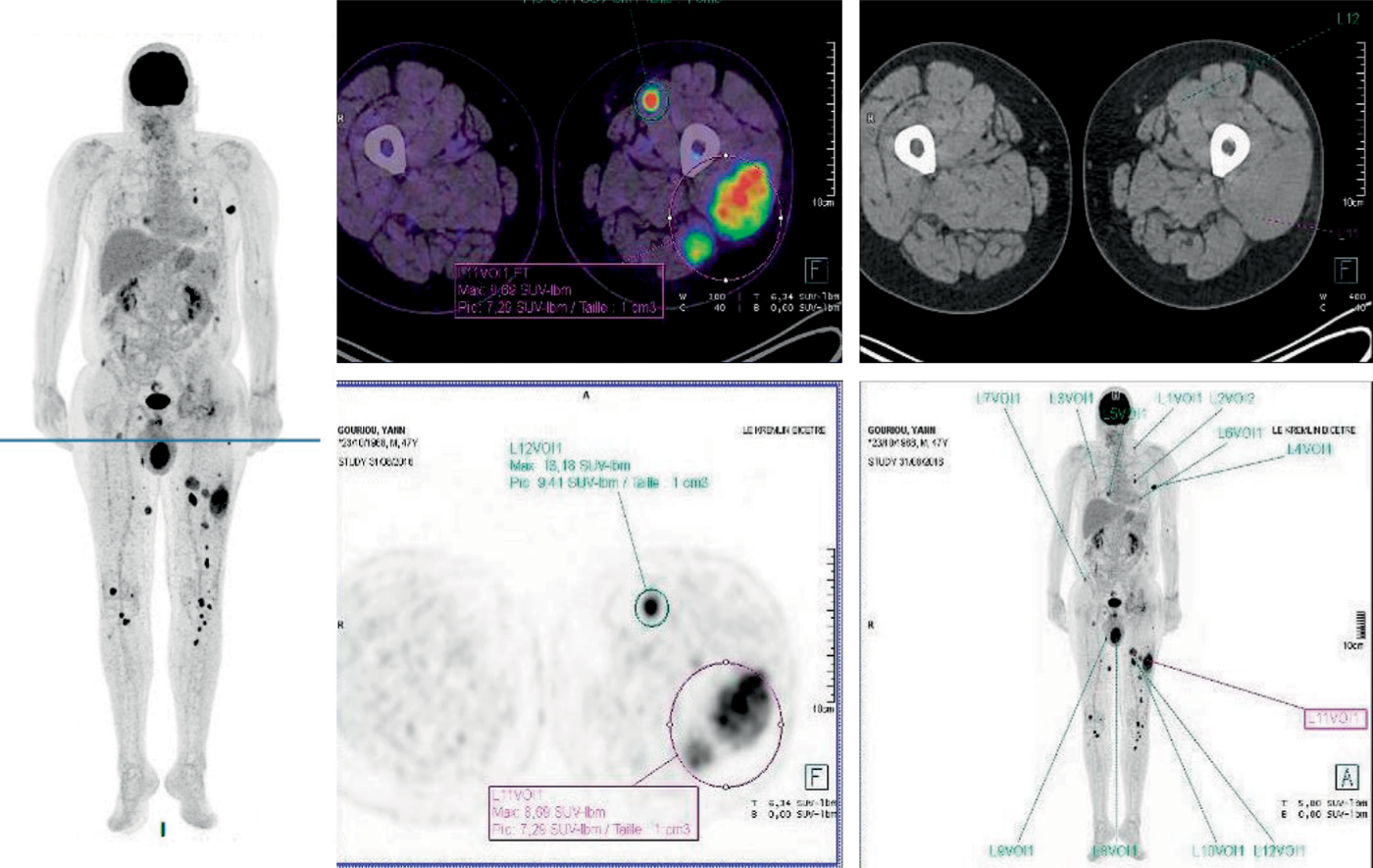
![]() Le TEP-scan est plus sensible que les radiographies standards :
Le TEP-scan est plus sensible que les radiographies standards :
Fig. 28.5. ![]() Image « corps entier » d’un TEP-scan. Patient atteint de myélome multiple en rechute sous forme extramédullaire
Image « corps entier » d’un TEP-scan. Patient atteint de myélome multiple en rechute sous forme extramédullaire
La scintigraphie osseuse n'a aucun intérêt : elle est le plus souvent normale ; les atteintes osseuses myélomateuses sont rarement hyperfixantes car ostéolytiques pures.
La prolifération plasmocytaire médullaire entraîne une raréfaction de l’hématopoïèse normale associant à des degrés divers anémie, thrombopénie, neutropénie.
L’anémie peut être liée à plusieurs mécanismes :
L’absence de plasmocytes normaux, secondaire à l’envahissement tumoral médullaire, peut entraîner une baisse de la synthèse des Ig normales. Ainsi, en cas de myélome, on a très souvent à la fois un pic monoclonal et une hypogammaglobulinémie polyclonale.
Il s’agit de fractures et de l’hypercalcémie. Les fractures vertébrales exposent au risque de compression médullaire et l’hypercalcémie à des complications cardiaques (troubles du rythme), neurologiques ou digestives (cf. item 268 au chapitre 26).
L’hypogammaglobulinémie polyclonale favorise les infections bactériennes récidivantes, en particulier ORL et pulmonaires.
Environ 50 % des patients atteints de myélome présentent une atteinte rénale ; celle-ci est due à la production excessive de chaînes légères d’Ig monoclonales. Il en existe cinq types.
Elle est fréquente : 80 % des insuffisances rénales aiguës au cours du myélome multiple sont liées à cette tubulopathie.
Elle est liée à la précipitation intratubulaire de chaînes légères d’immunoglobulines monoclonale. Elle s’observe plus souvent au cours des myélomes à chaînes légères et des myélomes à IgD.![]() La précipitation des cylindres est favorisée par :
La précipitation des cylindres est favorisée par :
L’utilisation de médicaments néphrotoxiques et de produits de contraste iodés est donc formellement contre-indiquée dans le myélome.
![]() La tubulopathie de type proximal avec syndrome de Fanconi est très rare ; elle est le témoin d’une toxicité particulière des chaînes légères kappa vis-à-vis des cellules tubulaires.
La tubulopathie de type proximal avec syndrome de Fanconi est très rare ; elle est le témoin d’une toxicité particulière des chaînes légères kappa vis-à-vis des cellules tubulaires.
Elle associe une tubulopathie avec glycosurie à glycémie normale, aminoacidurie généralisée, hypophosphatémie, acidose chronique.
L’hypercalcémie aiguë qui accompagne volontiers les myélomes avec atteinte osseuse sévère peut entraîner un syndrome polyuro-polydipsique sévère, susceptible de se compliquer d’une déshydratation extracellulaire et d’une hypovolémie. Cette hypovolémie peut se compliquer d’une insuffisance rénale fonctionnelle parfois révélatrice du myélome.
![]() Sa fréquence est variable : 5 à 10 %. Elle se rencontre essentiellement dans le myélome à chaînes légères lambda. Les localisations tissulaires, rénales, cardiaques, neuro-logiques, synoviales, et la sémiologie clinique sont celles décrites dans l’amylose AL.
Sa fréquence est variable : 5 à 10 %. Elle se rencontre essentiellement dans le myélome à chaînes légères lambda. Les localisations tissulaires, rénales, cardiaques, neuro-logiques, synoviales, et la sémiologie clinique sont celles décrites dans l’amylose AL.![]() L’amylose et la maladie des dépôts se caractérisent par une protéinurie glomérulaire faite majoritairement d’albumine. L’électrophorèse des protéines urinaires permet de faire la différence avec une néphropathie tubulaire liée à la précipitation de chaînes légères. Cliniquement, le tableau associe néphropathie glomérulaire avec protéinurie, voire syndrome néphrotique.
L’amylose et la maladie des dépôts se caractérisent par une protéinurie glomérulaire faite majoritairement d’albumine. L’électrophorèse des protéines urinaires permet de faire la différence avec une néphropathie tubulaire liée à la précipitation de chaînes légères. Cliniquement, le tableau associe néphropathie glomérulaire avec protéinurie, voire syndrome néphrotique.
![]() La maladie des dépôts de chaînes légères, de chaînes lourdes ou d'immunoglobuline entière est également dénommée syndrome de Randall.
La maladie des dépôts de chaînes légères, de chaînes lourdes ou d'immunoglobuline entière est également dénommée syndrome de Randall.
À l’heure actuelle, la guérison du myélome ne peut pas être obtenue. L’objectif est d’atteindre une rémission la plus longue possible. Le traitement fait régresser la dysglobulinémie, corrige l’anémie, l’hypercalcémie, l’insuffisance rénale. L’os peut se reconstruire même si cela n’est pas le plus fréquent. Puis, la maladie évolue à nouveau.
Cependant, les progrès thérapeutiques ont permis d’allonger la survie. Chez le sujet âgé de moins de 65 ans, la survie médiane est d’environ 8 ans grâce à l’apport d’une phase de chimiothérapie à haute dose suivie d’autogreffe de cellules souches. Chez les patients plus âgés inéligibles à l’intensification suivie d’autogreffe, la survie médiane est moins bonne à 5 ans.
Le score pronostique international utilisé actuellement est la classification (ISS-révisé) fondée sur les taux de β2-microglobulinémie, d’albuminémie, de LDH et la présence d’anomalies cytogénétiques (tableau 28.2).
Tableau 28.2 : ![]() Score pronostique international (ISS 2005).
Score pronostique international (ISS 2005).
Source : Greipp PR, San Miguel J, Durie BGM, Crowley JJ, Barlogie B, Bladé J, et al. International staging system for multiple myeloma. J Clin Oncol 2005;23(15):3412–20.
| Stade I | Stade II | Stade III | |
|---|---|---|---|
| Critères |
β2-microglobuline < 3,5 mg/l et Albumine > 35 g/l et Taux de LDH normal et Pas de haut risque cytogénétique |
Ni I, ni III |
β2-microglobuline > 5,5 mg/l et Haut risque cytogénétique t(4;4), t(4;6), del17p ou Taux de LDH > N |
| Survie médiane | 62 mois | 45 mois | 29 mois |
![]() La surveillance est clinique et biologique. Elle a pour but d’apprécier l’efficacité et la tolérance du traitement et de dépister les rechutes du myélome.
La surveillance est clinique et biologique. Elle a pour but d’apprécier l’efficacité et la tolérance du traitement et de dépister les rechutes du myélome.
Le traitement doit permettre d’obtenir la disparition de la douleur et l’amélioration de l’état général. L’aggravation clinique doit faire rechercher une reprise évolutive de la pathologie.
La surveillance biologique comprendra :
![]() Le traitement antitumoral s’adresse aux myélomes symptomatiques (critères CRAB ou SLiM CRAB). Il a pour but de contrôler la prolifération des cellules myélomateuses.
Le traitement antitumoral s’adresse aux myélomes symptomatiques (critères CRAB ou SLiM CRAB). Il a pour but de contrôler la prolifération des cellules myélomateuses.
Le choix du traitement est adapté à l’âge (avant ou après 65 ans), au statut physiologique et aux comorbidités (fig. 28.6) ; la qualité de vie des patients doit être une priorité et ces stratégies permettent au patient de mener le plus souvent une vie normale :
L’objectif thérapeutique est d’obtenir une rémission complète ou une très bonne réponse partielle définie selon les critères de l’IMWG et traduit par l’HAS en 2010. Elles sont associées à une meilleure survie sans progression.
Cette réponse thérapeutique est jugée sur la disparition des signes cliniques et la réduction des anomalies biologiques, en particulier du taux de la protéine monoclonale sérique et/ou urinaire. La réponse complète se définit par la normalisation de la moelle osseuse (< 5 %).
Le but du traitement est d’obtenir la plus faible maladie résiduelle (on parle de « Minimal Residual Disease negative » ou MRD négative) évaluée sur la moelle osseuse. Celle-ci est corrélée à une meilleure survie globale et à une survie sans progression prolongée.
Fig. 28.6 :![]() Stratégie thérapeutique pour la prise en charge du myélome en première ligne.
Stratégie thérapeutique pour la prise en charge du myélome en première ligne.
Adapté de : Dimopoulos MA, Moreau P, Terpos E, Mateos MV, Zweegman S, Cook G, et al. Multiple myeloma: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2021;32(3):309–22.
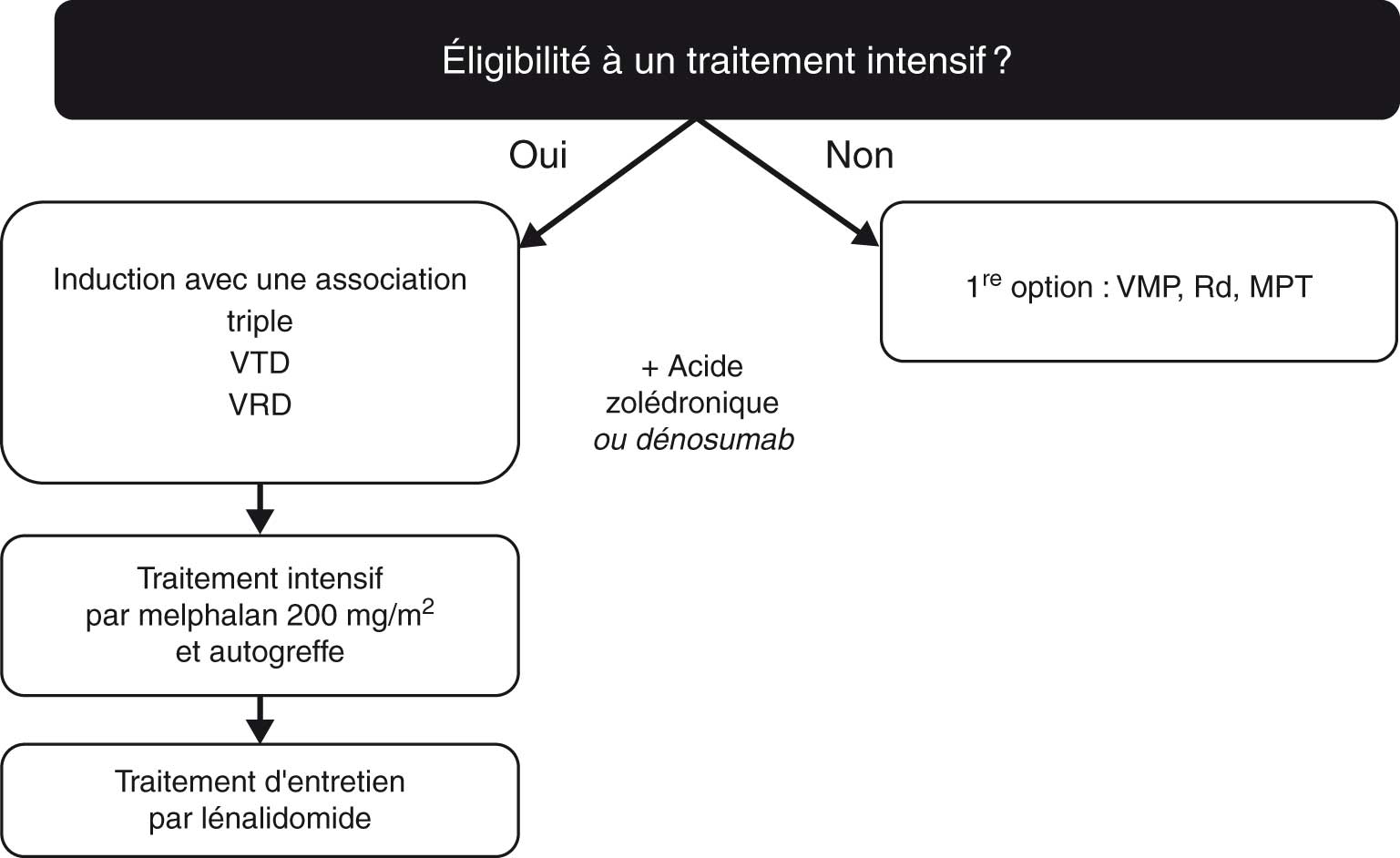
Les bisphosphonates réduisent l’incidence des complications osseuses (douleur, fracture, hypercalcémie) ; parallèlement, ils améliorent la qualité de vie.
On utilise préférentiellement l’acide zolédronique en perfusion mensuelle.
L’effet indésirable principal est l’ostéonécrose de la mâchoire et nécessite un bilan bucco-dentaire préalable (excepté si hypercalcémie symptomatique). Ce risque augmente de manière significative avec la durée de traitement et la dose cumulée.
Il est recommandé de traiter les patients nouvellement diagnostiqués avec atteinte osseuse pendant au moins 1 an.
Le dénosumab a aussi une autorisation de mise sur le marché dans cette indication, en cas d’impossibilité de l’acide zolédronique mais il expose au même risque d’ostéonécrose de la mâchoire. Il n’est cependant pas remboursé en France dans cette indication.
La radiothérapie est utilisée :
La cimentoplastie (introduction percutanée de ciment liquide par injection directe) ou la kyphoplastie (introduction de ciment à forte viscosité après dilatation par des ballons intravertébraux) sont particulièrement indiquées chez les patients présentant des douleurs rachidiennes avec ou sans fracture. Ils peuvent également stabiliser une lésion préfracturaire.
En l’absence de douleur sévère, la vertébroplastie est recommandée en cas de diminution significative de la hauteur, de l’intégrité structurale ou de la stabilité des corps vertébraux.
Le traitement chirurgical peut être indiqué :
![]() Il s’agit d’une lésion plasmocytaire unique, majoritairement osseuse. Le caractère unique doit être confirmé par un TEP-scan. Un composant monoclonal sanguin ou urinaire peut être objectivé. Le myélogramme est normal et les examens d’imagerie ne retrouvent pas d’autres lésions osseuses. Son traitement repose sur de la radiothérapie seule. En l’absence de traitement complètement éradicateur, le plasmocytome solitaire évolue habituellement vers un myélome, quelquefois sous la forme d’un plasmocytome multifocal, dont le suivi et le traitement sont les mêmes que pour un myélome multiple
Il s’agit d’une lésion plasmocytaire unique, majoritairement osseuse. Le caractère unique doit être confirmé par un TEP-scan. Un composant monoclonal sanguin ou urinaire peut être objectivé. Le myélogramme est normal et les examens d’imagerie ne retrouvent pas d’autres lésions osseuses. Son traitement repose sur de la radiothérapie seule. En l’absence de traitement complètement éradicateur, le plasmocytome solitaire évolue habituellement vers un myélome, quelquefois sous la forme d’un plasmocytome multifocal, dont le suivi et le traitement sont les mêmes que pour un myélome multiple
![]() La maladie de Waldenström est définie par :
La maladie de Waldenström est définie par :
Elle peut associer une hypertrophie ganglionnaire, splénique ou hépatique, des signes généraux (amaigrissement, sueurs), une anémie de mécanismes divers.
L'Ig peut être responsable d'un syndrome d'hyperviscosité avec céphalées, vertiges, d'une activité auto-immune (cryoglobulinémie, anti-MAG avec neuropathie, hémolyse).
![]() (Cf. item 318 dédié.)
(Cf. item 318 dédié.)
Il s'agit d'une prolifération monoclonale de lymphocytes B matures, de siège médullaire et sanguin. Les lymphocytes sont le plus souvent normaux morphologiquement, mais anormaux au plan fonctionnel.![]() Cette pathologie se caractérise par :
Cette pathologie se caractérise par :
Par ailleurs, il existe parfois :
![]() Plus rarement, l'immunoglobuline monoclonale révèle un lymphome malin non hodgkinien B ou une amylose.
Plus rarement, l'immunoglobuline monoclonale révèle un lymphome malin non hodgkinien B ou une amylose.
Légende :
Dans le respect de la Réforme du deuxième cycle des études médicales (R2C), les connaissances rassemblées sur ce site sont hiérarchisées en rang A, rang B et rang C à l'aide de balises et d'un code couleur :
![]() Connaissances fondamentales que tout étudiant doit connaître en fin de deuxième cycle.
Connaissances fondamentales que tout étudiant doit connaître en fin de deuxième cycle.
![]() Connaissances essentielles à la pratique mais relevant d'un savoir plus spécialisé que tout interne d'une spécialité doit connaître au premier jour de son DES.
Connaissances essentielles à la pratique mais relevant d'un savoir plus spécialisé que tout interne d'une spécialité doit connaître au premier jour de son DES.
![]() Connaissances spécifiques à un DES donné (troisième cycle).
Connaissances spécifiques à un DES donné (troisième cycle).
