Artérite à cellules géantes
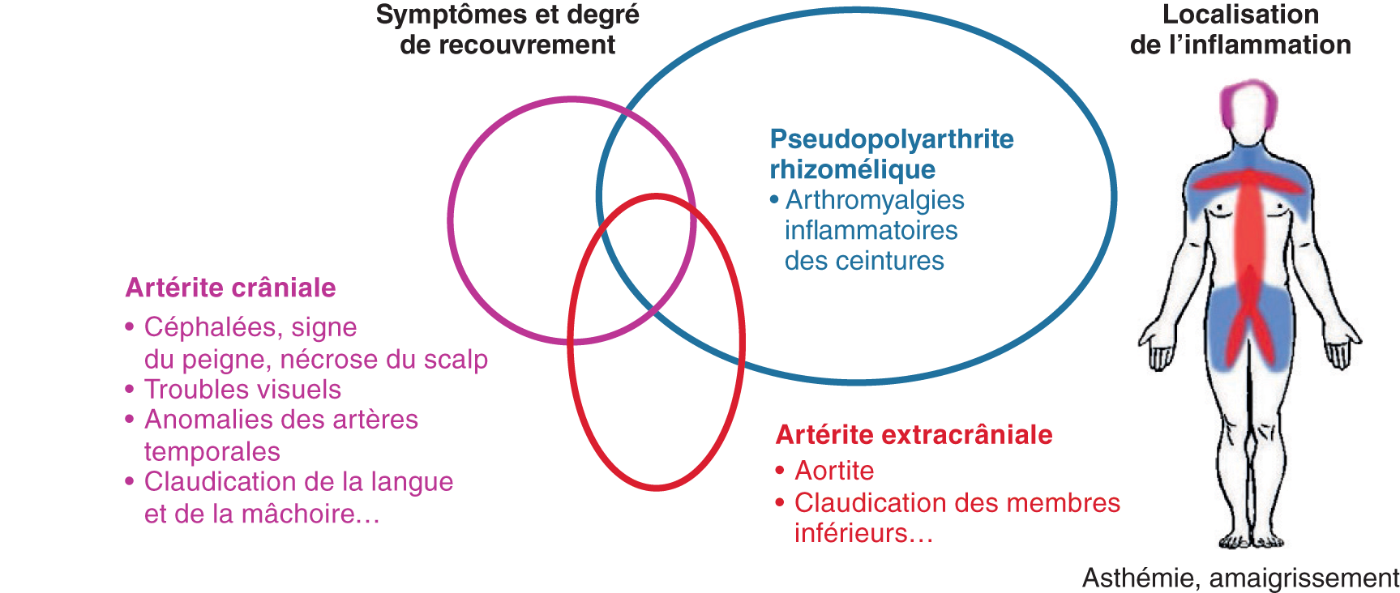
Fig. 16.1. ![]() Pseudopolyarthrite rhizomélique (PPR) et artérite à cellules géantes (ACG, ou maladie de Horton) crânienne et extracrânienne.
Pseudopolyarthrite rhizomélique (PPR) et artérite à cellules géantes (ACG, ou maladie de Horton) crânienne et extracrânienne.
![]() La physiopathologie de la PPR et de l’ACG n’est que partiellement comprise. Les principales hypothèses sont :
La physiopathologie de la PPR et de l’ACG n’est que partiellement comprise. Les principales hypothèses sont :
La mise en évidence d’une production importante d’IL-6 dans ces deux maladies a con- duit à l’évaluation d’anticorps monoclonaux inhibiteurs du récepteur de l’IL-6 dans les deux maladies, avec une efficacité démontrée (cf. section VI.E.2).
La PPR est un rhumatisme inflammatoire touchant le sujet âgé de plus de 50 ans, se ca- ractérisant cliniquement par des arthromyalgies (douleurs articulaires et des muscles) in- flammatoires (avec réveils nocturnes et dérouillage matinal prolongé) des ceintures (de rhiza, « racine » et mélos, « membre », qui désigne la localisation préférentielle à la ra- cine des membres) scapulaire et pelvienne. Elle se manifeste biologiquement par un syndrome inflammatoire.
Diagnostic de PPR
Le diagnostic de PPR repose sur des éléments clinico-biologiques évocateurs (douleurs inflammatoires des ceintures et CRP élevée) mais non spécifiques.
Il est en conséquence toujours nécessaire d’éliminer ses principaux diagnostics diffé- rentiels (cf. infra).
La constatation d’un tableau clinique compatible avec une PPR doit systématiquement faire rechercher cliniquement une ACG associée.
La PPR associe chez un patient de plus de 50 ans, après un début parfois insidieux :
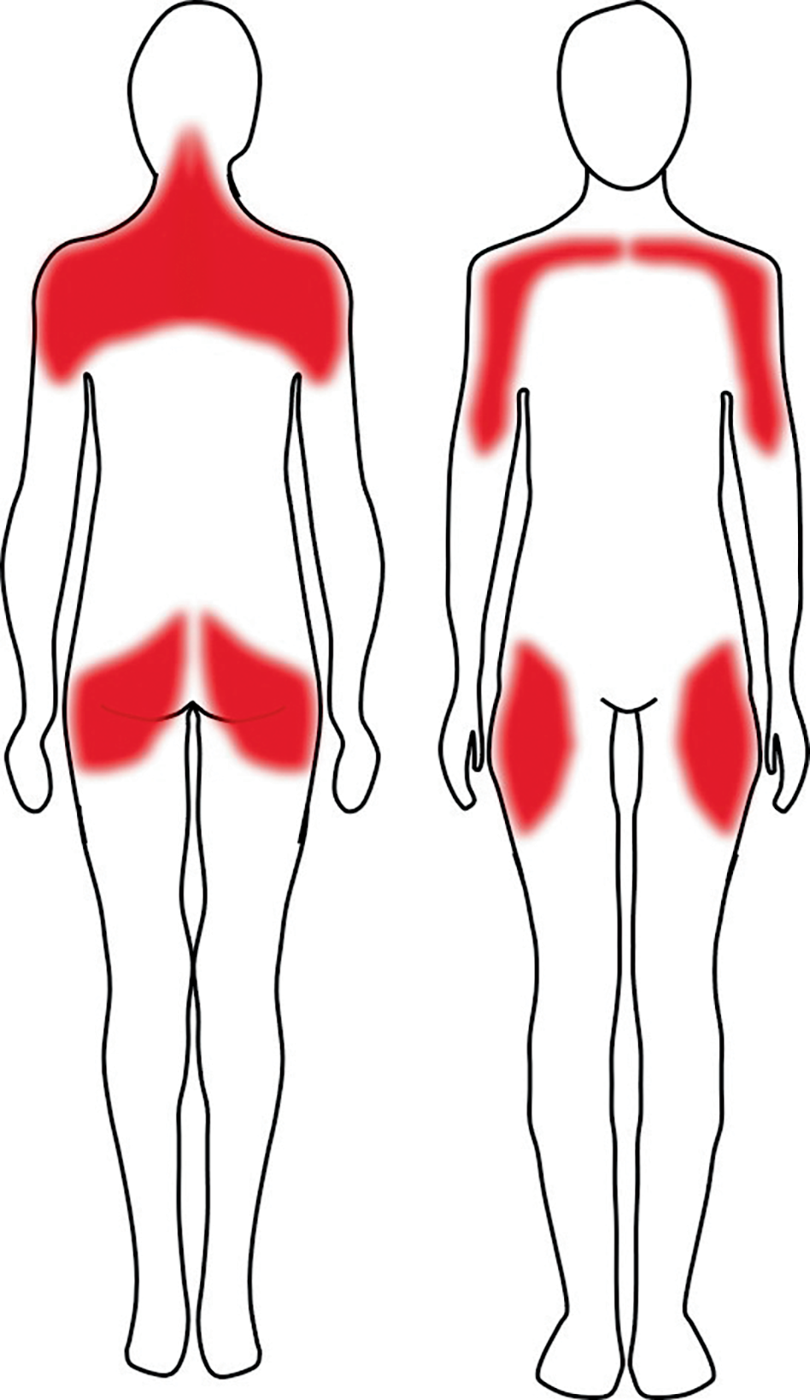
Fig. 16.2. ![]() Zones douloureuses au cours des arthromyalgies inflammatoires de la pseudopolyarthrite rhizomélique.
Zones douloureuses au cours des arthromyalgies inflammatoires de la pseudopolyarthrite rhizomélique.
![]() Il n’y a pas d’examens d’imagerie révélant des signes pathognomoniques de la PPR.
Il n’y a pas d’examens d’imagerie révélant des signes pathognomoniques de la PPR.
Des radiographies des articulations douloureuses doivent être réalisées, en particulier à la recherche d’un diagnostic différentiel (radiographies des épaules de face avec trois ro- tations et de profil, radiographie du bassin de face en charge).
Il n’y a pas de destruction articulaire dans la PPR.
Elle peut révéler (fig. 16.3) :
Dans la mesure où ces anomalies morphologiques sont isolément fréquentes chez le su- jet avançant en âge, c’est surtout le caractère bilatéral et inflammatoire (mode doppler) de ces anomalies qui oriente vers une PPR.
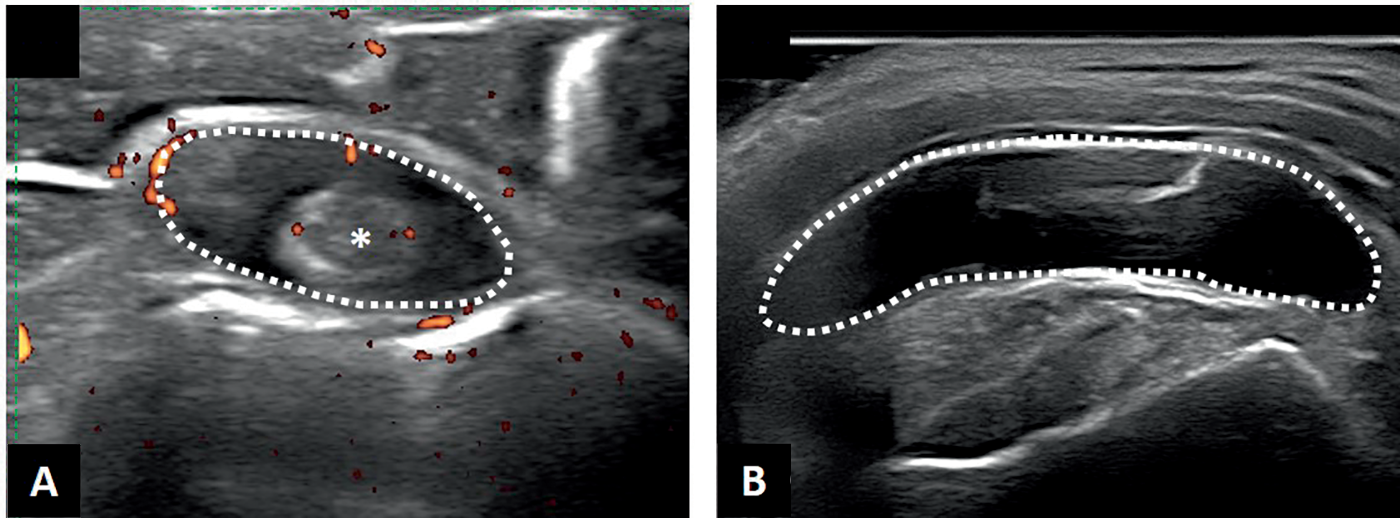
Fig. 16.3. ![]() Échographie dans la pseudopolyarthrite rhizomélique.
Échographie dans la pseudopolyarthrite rhizomélique.
A. Coupe transversale du long biceps (*) montrant une ténosynovite (tirets). B. Bursite sub-acromio-deltoïdienne (tirets).
Cet examen avec un marquage au 18-fluorodésoxyglucose (18FDG) permet de visualiser les zones métaboliquement actives consommant du glucose.
Il existe dans la PPR des fixations très importantes et bilatérales (même sur les sites asymptomatiques cliniquement) (fig. 16.4) :
Du fait de son coût, de son caractère irradiant et en accord avec les recommandations pour la prise en charge de la pseudopolyarthrite rhizomélique de la Société française de rhumatologie, cet examen doit être réservé à une prescription ciblée en cas de doute diagnostique.
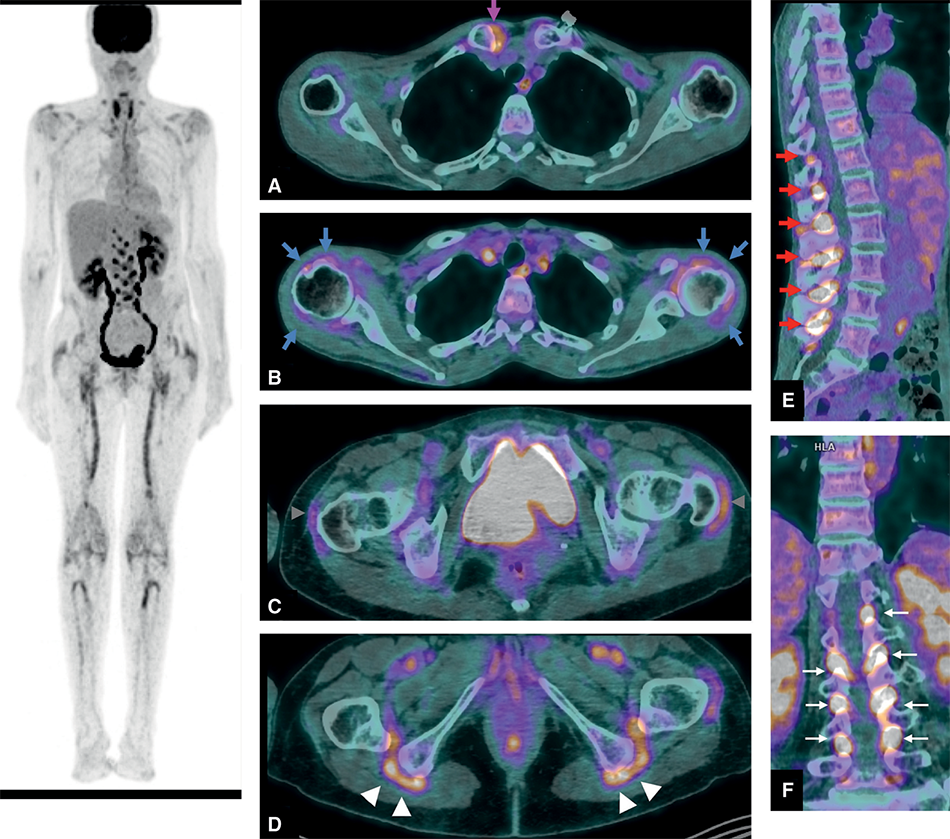
Fig. 16.4. ![]() TEP-scan dans la PPR.
TEP-scan dans la PPR.
Hypermétabolismes articulaires et péri-articulaires intenses prédominant sur les ceintures et touchant notamment l’articulation acromioclaviculaire droite (A, flèche rose), péri-articulaires des deux épaules (B, flèches bleues), des enthèses sur les grands trochanters (C, têtes de flèches grises) et les tubérosités ischiatiques (D, têtes de flèches blanches) ainsi que des es- paces interépineux (E et F, flèches rouges et blanches). À noter : un hypermétabolisme des artères fémorales, évocateur d’artérite à cellules géantes associée (astérisque).
Selon les recommandations de la Société française de rhumatologie pour la prise en charge de la PPR, en cas de présentation clinique typique, les seuls examens d’imagerie nécessaires au diagnostic sont les radiographies standards des sites atteints. Les autres examens complémentaires (tableau 16.1) sont adaptés en fonction des patients et des signes cliniques.
Tableau 16.1. ![]() Examens complémentaires (liste non exhaustive) à réaliser devant une suspicion de PPR.
Examens complémentaires (liste non exhaustive) à réaliser devant une suspicion de PPR.
| Examens systématiques | En fonction du contexte | Bilan préthérapeutique* | |
|---|---|---|---|
| Biologie | – Hémogramme – CRP – Ionogramme sanguin, calcémie – Urée, créatininémie et DFG – γGT, ASAT, ALAT – Anticorps anti-peptides citrullinés (anti-CCP) + facteurs rhumatoïdes |
– CPK, dosage « myosites » – TSH – Électrophorèse des protéines sériques – Anticorps antinucléaires, ANCA |
– Glycémie à jeun – Exploration d’une anomalie lipidique – Bandelette urinaire |
| Imagerie | – Radiographie des épaules (face, rotations neutre, interne et externe et profil) – Radiographie du bassin de face |
– Radiographie thoracique – Échographies des épaules et des hanches – TEP-scanner |
– Ostéodensitométrie |
| * Hors examens déjà réalisés. | |||
Le diagnostic de PPR repose sur un faisceau d’arguments positifs (arthromyalgies in- flammatoires des ceintures associées à une CRP élevée) et sur des arguments négatifs (absence d’arguments cliniques, biologiques et radiologiques en faveur d’une affection pouvant mimer une PPR) (tableau 16.2).
Tableau 16.2. ![]() Principaux diagnostics différentiels de la pseudopolyarthrite rhizomé- lique et points d’appels cliniques et biologiques de ces diagnostics.
Principaux diagnostics différentiels de la pseudopolyarthrite rhizomé- lique et points d’appels cliniques et biologiques de ces diagnostics.
| Catégorie | Diagnostic différentiel | Points d’appels cliniques | Points d’appels paracliniques |
|---|---|---|---|
| Autres rhumatismes inflammatoires | Rhumatisme à pyrophosphate de calcium et rhumatisme à hydroxyapatite | – Atteinte périphérique associée au poignet ou au genou – Début très brutal et intense, résolution progressive spontanée |
– Liseré de chondrocalcinose ou calcifications articulaires en radiographie ou échographie – Présence de microcristaux de pyrophosphate de calcium à la ponction articulaire |
| Polyarthrite rhumatoïde du sujet âgé | – Nombreuses arthrites périphériques et symétriques | – Anticorps anti-peptides cycliques citrullinés (anti-CCP) – Érosions typiques sur les radiographies |
|
| Remitting Seronegative Symetrical Synovitis with Pitting Edema (RS3PE) | – Œdème des extrémités prenant le godet | ||
| Spondyloarthrites du sujet âgé (rare) | – Atteinte rachidienne importante – Atteinte des articulations sacro-iliaques |
||
| Autres pathologies inflammatoires avec signes d’accompagnement | Myopathies inflammatoires | – Déficit musculaire | – Augmentation de la créatinine kinase – Présence d’autoanticorps des myopathies inflammatoires |
| Connectivites (rares) | – Lupus érythémateux systémique – Syndrome de Sjögren |
– Présence d’anticorps antinucléaires | |
| Cancers solides et hémopathies Iatrogène |
– Altération de l’état général prononcée – Fièvre – Rhumatismes inflammatoires induits (inhibiteurs des points de contrôle cellulaires) |
– Radiographie thoracique – TEP-scanner (en cas de forte suspicion) anormaux |
|
| Autres pathologies sans syndrome inflammatoire | Pathologies mécaniques | – Tendinopathie de la coiffe des rotateurs – Coxarthrose – Gonarthrose |
– CRP normale (le diagnostic de PPR ne doit pas être retenu en l’absence de syndrome inflammatoire) |
| Iatrogène | – Myalgies (statines, bêtabloquants, antiparkinsoniens) | ||
| Maladie de Parkinson | – Trouble de la marche – Syndrome extrapyramidal |
||
| Dysthyroïdies | – Goitre – Exophtalmie… |
||
| Ostéomalacies | – Douleurs et déformations osseuses… |
Le rhumatisme à pyrophosphate de calcium (RPPC) et le rhumatisme à hydroxyapatite (cf. item 198 au chapitre 19) sont les diagnostics différentiels les plus fréquents de la PPR. Le diagnostic différentiel entre PPR et polyarthrite rhumatoïde (cf. item 196 au chapitre 17) du sujet âgé est difficile car la polyarthrite rhumatoïde du sujet âgé a fréquemment un début rhizomélique (20 à 30 %) et la PPR peut comporter des ténosynovites et des arthrites périphériques (20 % des cas). La polyarthrite œdémateuse (RS3PE, Remitting Seronegative Symetrical Synovitis with Pitting Edema) comporte un œdème des extrémités, important, blanc, prenant le godet et une polysynovite. La polyarthrite œdémateuse est résolutive généralement en 12 à 18 mois. Elle est très corticosensible.
![]() L’artérite à cellules géantes (ACG, anciennement dénommée maladie de Horton) est une vascularite systémique primitive correspondant à une panartérite (inflammation attei- gnant toute la paroi des artères), segmentaire et focale des artères de grand et moyen ca- libre. Elle peut atteindre tous les vaisseaux de ces calibres, mais préférentiellement et par ordre de fréquence les branches de la carotide externe (artères temporales, maxillaires, occipitales) ainsi que les artères ophtalmiques, vertébrales, subclaviculaires, axillaires et l’aorte thoracique. On décrit classiquement les formes crâniales qui sont les plus fréquentes et les formes extracrâniales.
L’artérite à cellules géantes (ACG, anciennement dénommée maladie de Horton) est une vascularite systémique primitive correspondant à une panartérite (inflammation attei- gnant toute la paroi des artères), segmentaire et focale des artères de grand et moyen ca- libre. Elle peut atteindre tous les vaisseaux de ces calibres, mais préférentiellement et par ordre de fréquence les branches de la carotide externe (artères temporales, maxillaires, occipitales) ainsi que les artères ophtalmiques, vertébrales, subclaviculaires, axillaires et l’aorte thoracique. On décrit classiquement les formes crâniales qui sont les plus fréquentes et les formes extracrâniales.
Diagnostic d’ACG
Le diagnostic d’ACG repose sur l’association de signes cliniques évocateurs (signes de vascularite ou signes de PPR), d’un syndrome inflammatoire biologique et d’une preuve de vascularite (en imagerie ou sur une biopsie d’artère temporale).
(Cf. supra.)
Il est présent chez 15 % des malades et parfois révélateur d’ACG.
L’altération de l’état général et la fièvre sont habituellement plus marquées que dans la PPR isolée.
Urgence thérapeutique
Les signes d’urgence thérapeutique que sont les symptômes vasculaires oculaires sont à rechercher systématiquement :
Le moindre signe ophtalmologique doit faire redouter une complication ophtalmique grave et définitive et faire discuter une corticothérapie en urgence.
Fig. 16.5. ![]() Artères des territoires carotidiens internes (orange), carotidiens externes (rouge) et vertébraux (violet) classiquement atteintes dans l’artérite à cellules géantes et leurs manifestations cliniques associées.
Artères des territoires carotidiens internes (orange), carotidiens externes (rouge) et vertébraux (violet) classiquement atteintes dans l’artérite à cellules géantes et leurs manifestations cliniques associées.
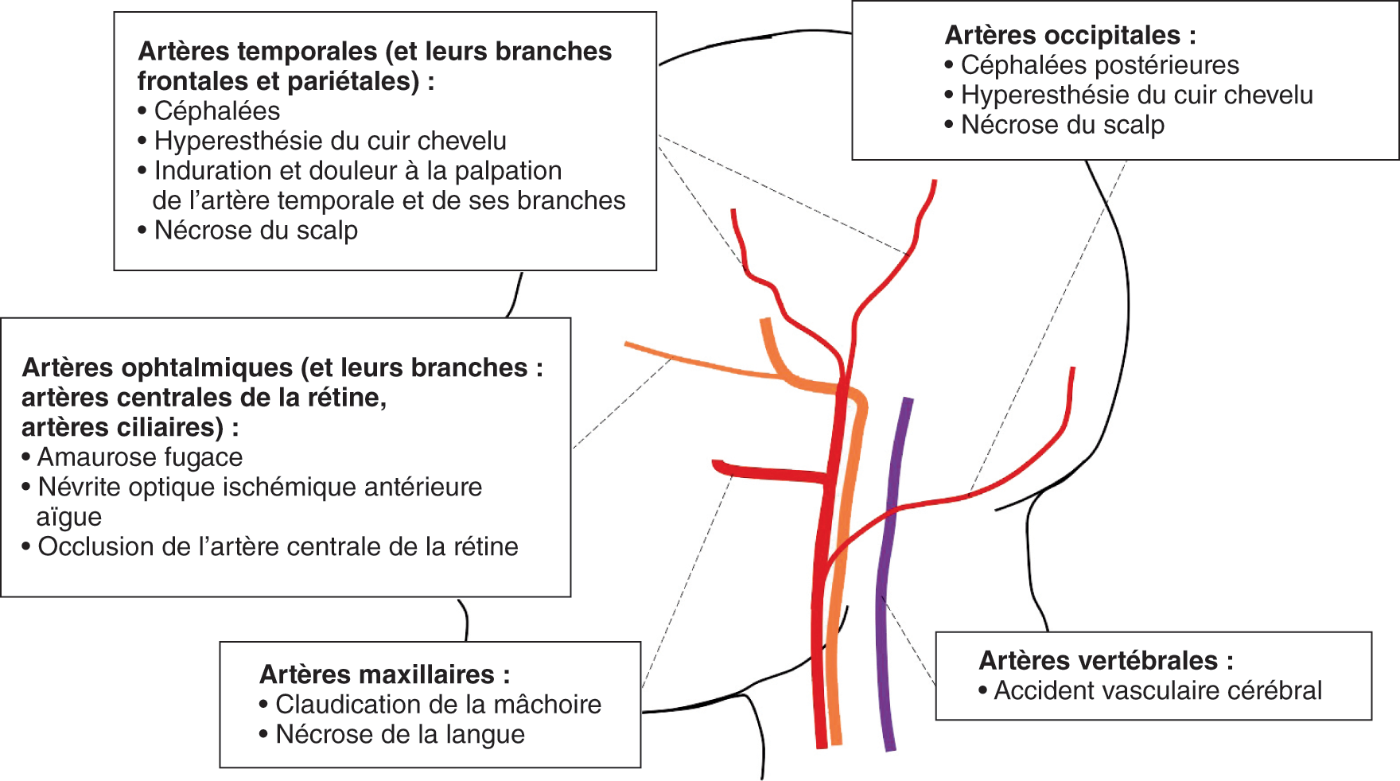
Les manifestations les plus sévères de l’ACG sont essentiellement vasculaires et isché- miques. Elles sont le plus souvent brutales et irréversibles. Elles font toute la gravité de la ma- ladie et doivent être recherchées systématiquement lors du diagnostic et des consultations de suivi.
Manifestations oculaires
Une complication oculaire se révèle le plus souvent par une cécité monoculaire brutale pouvant être précédée de prodromes (flou visuel, scotome, diplopie) : 1 à 2 % des pa- tients ont une cécité bilatérale définitive et 2 à 5 % une cécité monoculaire.
L’amaurose est la conséquence :
En cas d’atteinte unilatérale, le risque d’atteinte controlatérale et de cécité totale définitive est important et justifie une prise en charge thérapeutique urgente.
Complications neurologiques
Autres complications
![]() C’est l’examen de première intention en cas de suspicion d’ACG. L’échographie des axes vasculaires, notamment des artères temporales, peut révéler des signes caractéris- tiques d’artérite. Il est recommandé d’étudier également les gros vaisseaux du cou et les troncs supra-aortiques (artères carotides et axillaires).
C’est l’examen de première intention en cas de suspicion d’ACG. L’échographie des axes vasculaires, notamment des artères temporales, peut révéler des signes caractéris- tiques d’artérite. Il est recommandé d’étudier également les gros vaisseaux du cou et les troncs supra-aortiques (artères carotides et axillaires).
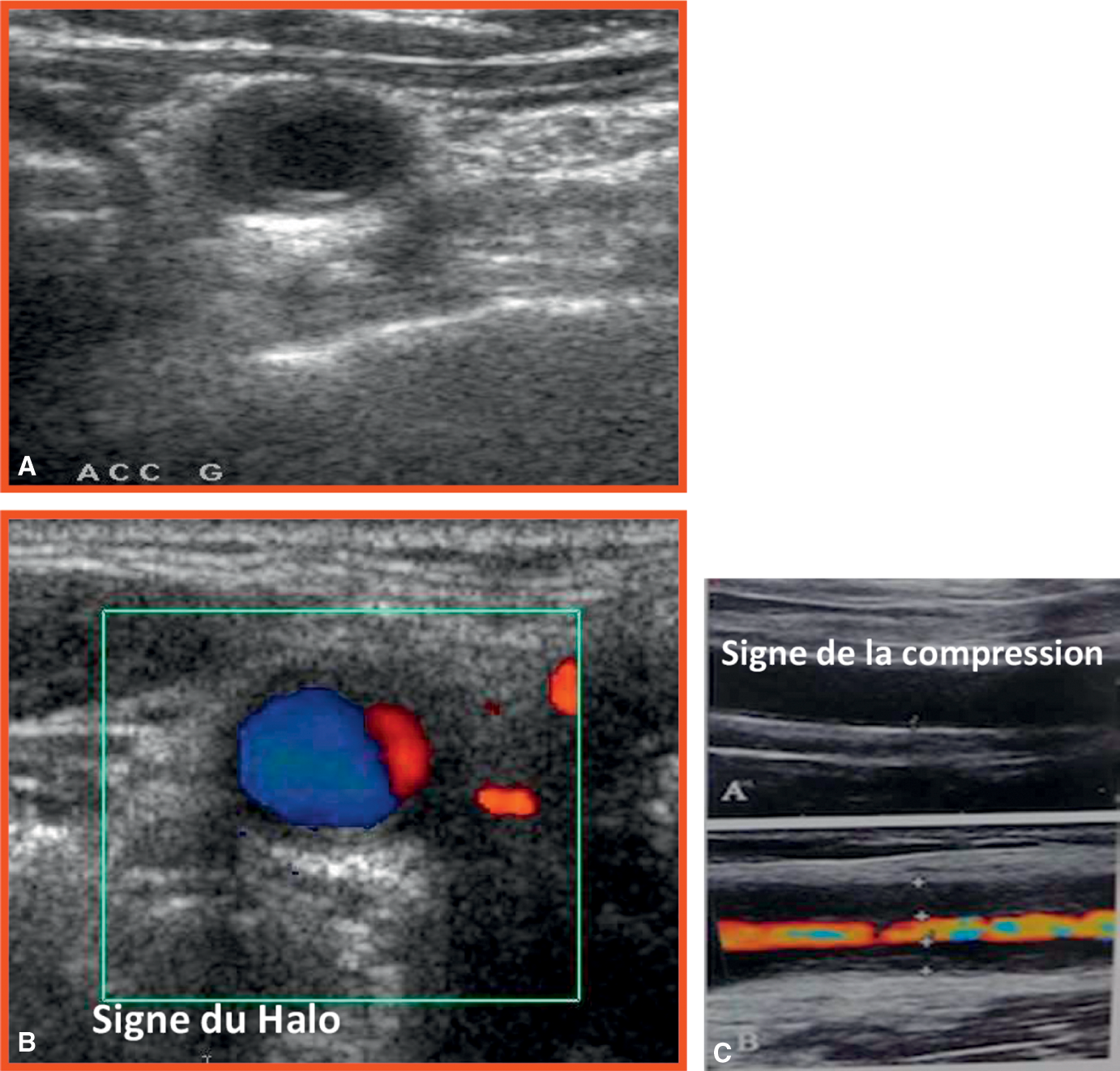
Le signe pathognomonique est le signe du halo (halo hypoéchogène circonférentiel de la paroi du vaisseau). Une incompressibilité de l’artère, une occlusion ou une sté- nose peuvent aussi être observées. L’échographie (fig. 16.6) doit impérativement être réalisée par un opérateur entraîné sachant faire la différence entre un signe du halo et des lésions athéromateuses, fréquentes à cet âge. Cet examen peut donc per- mettre de confirmer le diagnostic ou de diriger la biopsie de l’artère afin d’améliorer la sensibilité diagnostique de cet examen.
La présence d’un signe du halo bilatéral a une spécificité de 100 % et permet devant un tableau clinico-biologique compatible de retenir le diagnostic d’ACG sans réaliser de biopsie d’artère temporale.
Signe du halo
Il consiste en un halo hypoéchogène circonférentiel au niveau de la paroi du vaisseau qui correspond à son inflammation, une hypoéchogénicité de l’intima du vaisseau touché par l’inflammation et, de manière moins spécifique, une sténose pouvant aller jusqu’à l’occlusion complète.
Fig. 16.6. ![]() Échographie mode B et doppler des artères temporales.
Échographie mode B et doppler des artères temporales.
A. Signe du halo en mode B. B. Signe du halo en mode doppler. C. Signe de la compression : en compression, la paroi de l’artère (hypoéchogène) reste visible, sans le flux doppler.
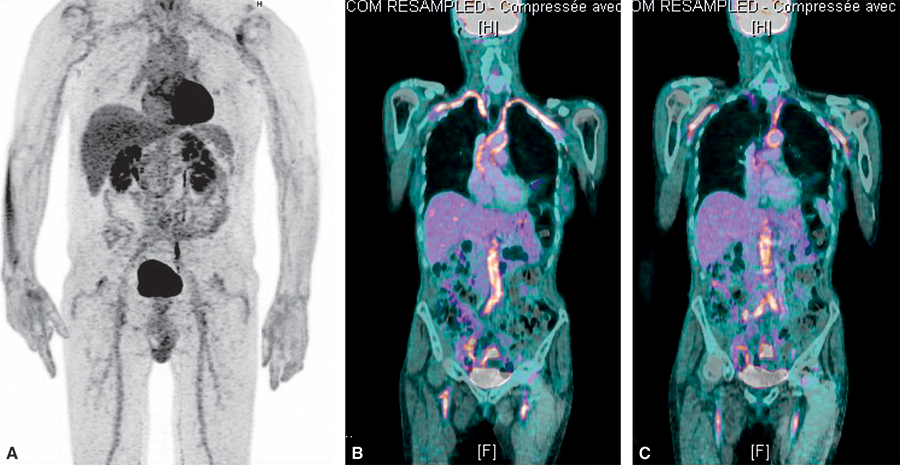
Cet examen au 18FDG, comme dans la PPR, permet de visualiser les atteintes des vais- seaux extracrâniens (fig. 16.7) et d’obtenir une cartographie de l’ensemble des vaisseaux atteints, montrant des zones inflammatoires de fixation intense du traceur radioactif utili- sé. C’est actuellement un des examens de référence dans le diagnostic de l’artérite à cel- lules géantes extracrâniale. Les évolutions techniques rapides de la TEP permettent maintenant d’étudier également les vaisseaux crâniens sur les appareils les plus modernes.
![]() Les coupes scanographiques associées permettent de visualiser plus précisément l’atteinte de l’aorte. Son utilisation pourrait permettre d’évaluer l’activité de la maladie et constituerait un outil intéressant pour le suivi. Il n’est cependant pas réalisé de manière systématique.
Les coupes scanographiques associées permettent de visualiser plus précisément l’atteinte de l’aorte. Son utilisation pourrait permettre d’évaluer l’activité de la maladie et constituerait un outil intéressant pour le suivi. Il n’est cependant pas réalisé de manière systématique.
![]() Le diagnostic histologique est, du fait des progrès de l’imagerie, de moins en moins nécessaire au diagnostic.
Le diagnostic histologique est, du fait des progrès de l’imagerie, de moins en moins nécessaire au diagnostic.
Fig. 16.7. ![]() TEP-scanner dans l’artérite à cellules géantes.
TEP-scanner dans l’artérite à cellules géantes.
Hyperfixations très évocatrices d’une artérite au niveau des artères axillaires (flèches blanches), de l’aorte abdominale (flèches rouges) et des artères fémorales (flèches jaunes).
![]() Ces deux examens sont utiles au diagnostic des atteintes extracrâniennes de l’ACG et font partie des examens cités dans le protocole national de soin de la HAS actualisé en 2024. L’angioscanner est également utile au suivi de l’atteinte aortique et à la recherche de compli- cations.
Ces deux examens sont utiles au diagnostic des atteintes extracrâniennes de l’ACG et font partie des examens cités dans le protocole national de soin de la HAS actualisé en 2024. L’angioscanner est également utile au suivi de l’atteinte aortique et à la recherche de compli- cations.
![]() La biopsie de l’artère temporale (BAT) est un des examens de référence pour confir- mer une ACG. Mais le diagnostic peut également être réalisé grâce à l’imagerie dans les centres experts (imagerie de l’artère temporale ou de l’aorte et de ses branches), se substituant à la BAT.
La biopsie de l’artère temporale (BAT) est un des examens de référence pour confir- mer une ACG. Mais le diagnostic peut également être réalisé grâce à l’imagerie dans les centres experts (imagerie de l’artère temporale ou de l’aorte et de ses branches), se substituant à la BAT.
La BAT ne doit pas être réalisée devant un tableau de PPR isolée typique sans signes cliniques évocateurs d’atteinte vasculaire inflammatoire.
La BAT ne doit pas retarder le traitement. Les anomalies histologiques persistent au moins 15 jours après le début de la corticothérapie.
Le caractère focal de l’artérite justifie un prélèvement de 1 cm de long ou plus. L’examen anatomopathologique met en évidence une panartérite gigantocellulaire segmentaire et focale.
Pour être considérée comme preuve histologique d’une ACG, la biopsie doit nécessaire- ment montrer un infiltrat inflammatoire mononucléé de la média et/ou de l’intima. La pré- sence surajoutée d’une destruction de la limitante élastique interne et/ou de cellules géantes est pathognomonique mais inconstante.
La négativité de la biopsie de l’artère temporale n’exclut pas le diagnostic d’ACG. Le ca- ractère focal de la maladie explique la fréquence des faux négatifs (sensibilité de la biop- sie de l’artère temporale : entre 50 et 91 %).
Dans les formes typiques d’ACG, il y a peu de diagnostics différentiels.
Elles sont plus rares à cet âge : maladie de Takayasu, qui atteint le sujet jeune, granulo- matose avec polyangéite, péri-artérite noueuse, maladie de Behçet.
Règles hygiéno-diététiques
Autres
![]() Le traitement d’attaque est la prednisone à 0,7–1 mg/kg par jour (40 à 80 mg par jour environ, sans dépasser 80 mg) à débuter assez rapidement pour éviter les complications ischémiques.
Le traitement d’attaque est la prednisone à 0,7–1 mg/kg par jour (40 à 80 mg par jour environ, sans dépasser 80 mg) à débuter assez rapidement pour éviter les complications ischémiques.
En cas d’ACG compliquée de signes oculaires ou ischémiques : des bolus de méthyl- prednisolone peuvent être proposés mais sans preuve d’une meilleure efficacité. Cer- taines équipes proposent d’associer un traitement antiagrégant préventif.
La décroissance progressive n’est proposée qu’après 2 à 4 semaines et disparition des signes cliniques et régression du syndrome inflammatoire biologique.
L’absence de corticosensibilité doit faire remettre en cause le diagnostic.
Le tocilizumab (anticorps monoclonal anti-IL-6R) est efficace dans le traitement de l’ACG et de la PPR. Il a obtenu une autorisation de mise sur le marché en 2017 dans l’ACG. Le sarilumab est efficace dans la PPR mais n’a pas d’autorisation de mise sur le marché en France.
La prescription des inhibiteurs du récepteur de l’IL-6 est réservée actuellement aux spé- cialistes du domaine, dans un but d’épargne cortisonique chez des patients ayant des comorbidités (ostéoporose fracturaire sévère, diabète décompensé, troubles psychiatriques).
Le méthotrexate permet une épargne en corticoïdes dans la PPR et l’ACG mais les preuves disponibles sont moins solides que pour les inhibiteurs du récepteur de l’IL-6.
![]() La maladie de Takayasu est une vascularite des gros vaisseaux touchant principale- ment les femmes (70 %) jeunes (20 à 35 ans) et atteignant préférentiellement l’aorte et ses gros troncs, ainsi que l’artère pulmonaire. Elle peut atteindre tous les gros vaisseaux mais dans 60 à 90 % des cas, il s’agit de l’aorte ascendante ou descendante, des artères subclaviculaires et des branches de la carotide. Elle était communément appelée la « maladie des femmes sans pouls ».
La maladie de Takayasu est une vascularite des gros vaisseaux touchant principale- ment les femmes (70 %) jeunes (20 à 35 ans) et atteignant préférentiellement l’aorte et ses gros troncs, ainsi que l’artère pulmonaire. Elle peut atteindre tous les gros vaisseaux mais dans 60 à 90 % des cas, il s’agit de l’aorte ascendante ou descendante, des artères subclaviculaires et des branches de la carotide. Elle était communément appelée la « maladie des femmes sans pouls ».
La maladie évolue habituellement selon deux phases.
Elle peut associer :
Cette phase dure de quelques semaines à quelques mois. Elle peut récidiver ou dispa- raître avec un temps de latence de plusieurs années avant la survenue de la phase oc- clusive.
Les signes cliniques sont liés à la sténose, l’occlusion ou l’anévrisme des artères et va- rient selon le territoire vasculaire atteint. Des formes longtemps indolentes ou avec des signes peu spécifiques peuvent aboutir à des retards de diagnostic.
Les signes les plus fréquents sont l’apparition d’une HTA, d’une asymétrie tensionnelle, la disparition d’un pouls (notamment huméral), la claudication d’un membre supérieur chez un sujet de moins de 40 ans (syndrome de l’arc aortique) :
Les accidents vasculaires cérébraux et les infarctus du myocarde sont rares.
Il n’y a aucun marqueur sérologique spécifique. Le syndrome inflammatoire (VS ou CRP) est non spécifique mais évocateur d’une artérite. Il peut être absent même en cas de ma- ladie active.
![]() La confirmation diagnostique se fait grâce à l’imagerie ou à l’anatomie pathologique.
La confirmation diagnostique se fait grâce à l’imagerie ou à l’anatomie pathologique.
![]() Actuellement ce sont l’échographie-doppler artérielle (par des opérateurs très entraînés), l’IRM et le TEP-scan qui sont au premier plan pour la confirmation diagnostique.
Actuellement ce sont l’échographie-doppler artérielle (par des opérateurs très entraînés), l’IRM et le TEP-scan qui sont au premier plan pour la confirmation diagnostique.
L’angioscanner est moins fréquemment utilisé.
![]() La biopsie artérielle montre des lésions inflammatoires multifocales et segmentaires touchant les trois tuniques. Elle peut être aspécifique ou montrer des lésions cicatri- cielles. Du fait des progrès de l’imagerie, le diagnostic histologique est de moins en moins nécessaire au diagnostic.
La biopsie artérielle montre des lésions inflammatoires multifocales et segmentaires touchant les trois tuniques. Elle peut être aspécifique ou montrer des lésions cicatri- cielles. Du fait des progrès de l’imagerie, le diagnostic histologique est de moins en moins nécessaire au diagnostic.
Légende :
Dans le respect de la Réforme du deuxième cycle des études médicales (R2C), les connaissances rassemblées sur ce site sont hiérarchisées en rang A, rang B et rang C à l'aide de balises et d'un code couleur :
![]() Connaissances fondamentales que tout étudiant doit connaître en fin de deuxième cycle.
Connaissances fondamentales que tout étudiant doit connaître en fin de deuxième cycle.
![]() Connaissances essentielles à la pratique mais relevant d'un savoir plus spécialisé que tout interne d'une spécialité doit connaître au premier jour de son DES.
Connaissances essentielles à la pratique mais relevant d'un savoir plus spécialisé que tout interne d'une spécialité doit connaître au premier jour de son DES.
![]() Connaissances spécifiques à un DES donné (troisième cycle).
Connaissances spécifiques à un DES donné (troisième cycle).
